
汪廣進1,2,程 凡1,徐 甜2,余 意2,文 勝1,龔春麗1,劉 海1,汪 杰1,鄭根穩1,潘 牧2
(1 湖北工程學院化學與材料科學學院,孝感432000;2 武漢理工大學材料復合新技術國家重點實驗室,湖北燃料電池重點實驗室,武漢430070)
摘 要 采用溶膠-凝膠法結合二次高溫燒結技術,制備了錳系鈣鈦礦催化劑。利用XRD和EDS對催化劑的物相與元素組成進行了分析,并利用電化學分析方法研究了催化劑的氧還原催化性能。XRD與EDS結果表明,N2氣氛二次燒結不改變La0.7Sr0.3-MnO3物相組成,但NH3氣氛二次燒結會造成La0.7Sr0.3MnO3分解。電化學結果表明,N2氣氛二次燒結催化劑的氧還原催化活性高于NH3氣氛二次燒結催化劑,其氧還原起始電勢與極限電流分別為0.028V(vs.Hg/HgO)和2.181mA·cm-2(2 000r/min)。
關 鍵 詞 二次燒結 La0.7Sr0.3MnO3 氧還原反應 催化活性
0 引言
目前,日益加劇的能源危機促進了新型可再生能源裝置的快速發展,特別是質子交換膜燃料電池(Proton exchangemembrane fuel cells,PEMFCs)[1]。PEMFCs的整體性能取決于其電催化劑的性能。目前已知催化活性最高的電催化劑是貴金屬鉑[2]。但貴金屬鉑成本高昂、資源稀缺,嚴重阻礙了PEMFCs的商業發展[3]。因此,非貴金屬電催化劑的研制對推動PEMFCs發展具有重要意義。
由于具有優異的物理化學性能,鈣鈦礦型過渡金屬氧化物備受PEMFCs電催化劑研究領域的關注[4-7]。本課題組前期研究發現酸性介質中La0.65Sr0.3MnO3粉末的氧還原起始電勢為0.50V(vs.SCE)[5],還發現堿性介質中650℃煅燒La1-xSrxMnO3樣品的氧還原起始電勢為0.127V(vs.Hg/HgO),高于750℃和850℃樣品[7]。Yang等[8]發現鈣鈦礦型過渡金屬氧化物的氧還原催化活性決定于eg軌道電子填充度與B 位原子含氧基團吸附強度,其中錳系氧化物(LaMn-O3)鈣鈦礦的氧還原催化活性最高,它的eg軌道電子填充度為1.0。采用第一性原理方法,Cheng等[9]發現LaMn-O3的氧還原電催化活性高于LaCrO3和LaFeO3,與Yang等[8]研究結果一致。Donne等[10]發現堿性介質中La1-x-SrxMnO3的氧還原起始電勢約為0.0V (vs.Hg/HgO),與商業Pt/C的氧還原催化活性相當,但其擴散電流密度仍遠低于商業Pt/C,還不能滿足PEMFCs運行要求。
此外,本課題組前期的研究還發現在NH3氣氛中950℃二次燒結處理燒綠石La2Zr2O7,可將其氧還原起始電勢、半波電勢和極限電流密度分別提高6.32%、6.02% 和8.27%[11]。因此,本實驗擬以鈣鈦礦La0.7Sr0.3MnO3為研究對象,采用溶膠-凝膠法結合二次高溫燒結技術,借助物理化學與電化學表征方法,研究二次燒結氣氛對La0.7Sr0.3MnO3氧還原催化活性的影響。
1 實驗
1.1 催化劑制備
本實驗所用化學試劑均為分析純級,購自國藥集團化學試劑有限公司。借鑒實驗室溶膠-凝膠法合成經驗[5-7],制備La0.7Sr0.3MnO3前驅體。首先,配制體積比為1∶1的乙醇水溶液。按照金屬離子物質的量比為0.7∶0.3∶1稱取La-(NO3)3·6H2O、Sr(NO3)2和Mn(NO3)2溶于混合溶液中,配制成硝酸鹽溶液。再加入與金屬離子物質的量比為2∶1的檸檬酸,形成檸檬酸硝酸鹽溶液。然后,在恒溫水浴鍋中80℃熱處理檸檬酸硝酸鹽溶液,將溶液變成溶膠。將所得溶膠置于真空干燥箱內80℃干燥12h,形成蜂窩狀蓬松干凝膠。接著,將獲得的干凝膠置于石英管式爐中650℃預煅燒3h,獲得La0.7Sr0.3MnO3前驅體。最后,在石英氣氛管式爐中,NH3和N2氣氛保護下,950℃二次燒結處理La0.7Sr0.3-MnO3前驅體24h獲得催化劑樣品。
1.2 催化劑物化性能表征
利用X射線衍射儀分析催化劑前驅體與催化劑的物相組成。其中,X射線源為Cu Kα,管電壓為40kV,管電流為50mA,掃描范圍為20~80°,掃描速度為5(°)/min,步長為0.02°,精度為Δ2θ≤±0.02°。利用掃描電子顯微鏡分析催化劑樣品的微觀形貌結構,并結合X射線能譜儀定量分析催化劑樣品的元素組成。
1.3 催化劑電化學性能表征
分別稱取一定量的催化劑樣品、碳粉和5%(質量分數)Nafion溶液,加入體積比為1∶1的乙醇/水溶液中,超聲分散處理30min,制得催化劑墨水,其中1mL催化劑墨水含5mg催化劑、1mg碳粉和20μL Nafion。然后,用微量移液器移取10μL催化劑墨水滴加到玻璃碳電極上,待溶劑完全揮發,制得工作電極,其中1cm2 工作電極表面含0.5mg催化劑和100μg碳粉。將工作電極浸入0.5mol/L KOH溶液待測。采用傳統三電極體系,分別以鉑電極和Hg/HgO電極為對電極和參比電極,評價催化劑樣品的氧還原催化活性。其中,循環伏安掃描和線性電勢掃描測試的掃描速率分別為20mV/s和5mV/s,電勢范圍為-0.6~0.2V(vs.Hg/HgO)。本實驗中所有電流密度均以玻璃碳電極幾何面積為催化活性面積進行歸一化,所有測試都在室溫下完成。
2 結果與討論
2.1 物相及形貌分析
圖1為La0.7Sr0.3MnO3前驅體的XRD圖譜。從圖1可以看出,前驅體氧化物的主要衍射峰峰位分別位于2θ =22.9°、32.9°、40.3°、47.0°、52.8°、58.7°、68.7°和78.0°,這些衍射峰峰位分別與鈣鈦礦型氧化物La0.7Sr0.3MnO3的(012)、(104)、(202)、(024)、(122)、(214)、(220)和(134)晶面衍射峰峰位對應。此外,沒有出現其他雜質衍射峰。結果表明,采用溶膠凝膠法可以合成出純度較高的La0.7Sr0.3MnO3鈣鈦礦型氧化物,與本課題組之前報道結果相符。
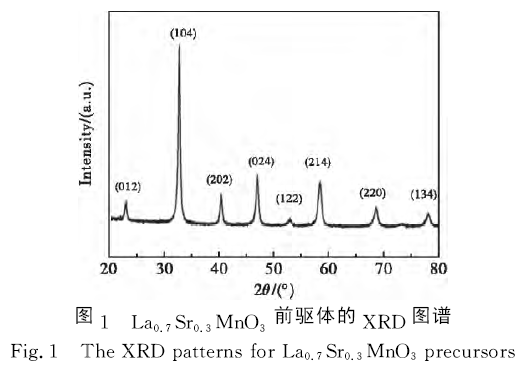
為分析二次燒結氣氛對催化劑物相的影響,圖2給出了在NH3和N2氣氛中二次燒結催化劑的XRD圖譜。從圖2可以看出,經N2氣氛二次燒結催化劑的XRD圖譜類似于La0.7Sr0.3MnO3前驅體XRD圖譜,說明經N2氣氛二次燒結不會改變La0.7Sr0.3MnO3鈣鈦礦物相結構。但經NH3氣氛二次燒結催化劑XRD圖譜完全不同于La0.7Sr0.3MnO3前驅體XRD圖譜,說明經NH3氣氛二次燒結破壞了La0.7Sr0.3-MnO3鈣鈦礦物相組成。Atsumi等[12]發現在1% H2還原氣氛中La1-σSrσMnO3(0.2≤σ≤0.5)高溫分解為(La1-x-Srx)2MnO4和MnO。Andrieux等[13]發現在極低O2濃度中La0.8Sr0.2MnO3±δ高溫分解為La2O3、SrMnO3、La2MnO4和MnO。對比分析發現,在NH3氣氛中二次燒結后,La0.7Sr0.3-MnO3前驅體部分分解為SrMnO2.694。與(La0.7Sr0.3)2MnO4和MnO的衍射峰峰位相比,分解產物衍射峰峰位均向左移動,說明分解產物晶格可能發生改變。
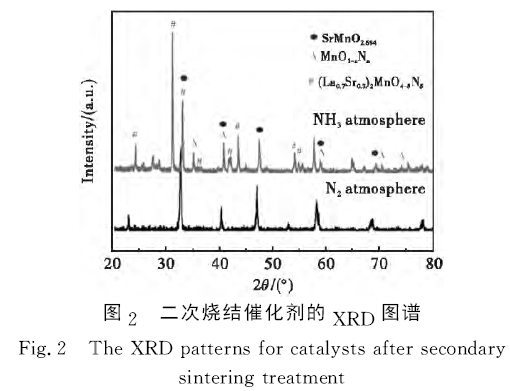
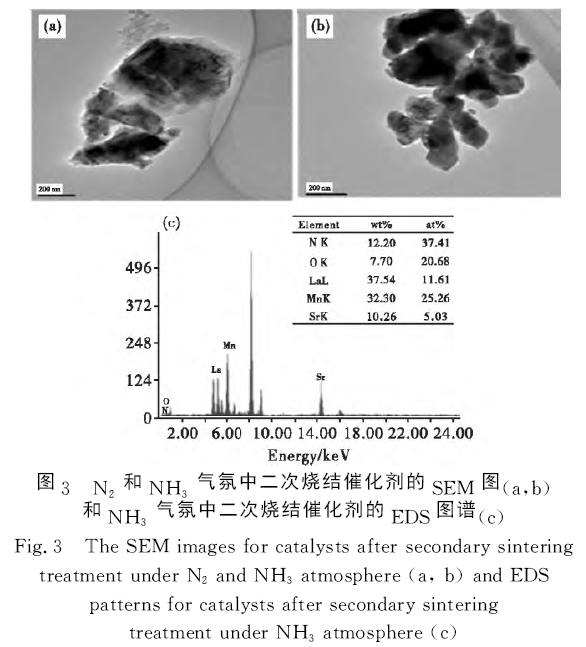
圖3(a)、(b)分別為N2和NH3氣氛中二次燒結催化劑的SEM 圖。由圖3(a)可以看出,N2氣氛中二次燒結催化劑顆粒粒徑約為250nm,這與二次燒結引起La0.7Sr0.3MnO3前驅體晶體再生長相關。相比N2氣氛中二次燒結催化劑的顆粒粒徑,NH3氣氛中二次燒結的部分催化劑顆粒粒徑約為100nm,可能是由于NH3氣氛中二次燒結會引起La0.7Sr0.3-MnO3前驅體的分解,從而造成前驅體晶體裂解。此外,圖3(c)為NH3氣氛中二次燒結催化劑的EDS圖譜。由圖3(c)可知,除元素La、Sr、Mn和O外,NH3氣氛中二次燒結催化劑中還含N元素。結合XRD與EDS分析可得,NH3氣氛中二次燒結前驅體分解為MnO1-xNx、(La0.7Sr0.3)2MnO4-δNδ(0≤δ<4)和SrMnO2.694。
2.2 電化學分析
圖4為碳粉與不同氣氛二次燒結催化劑樣品的循環伏安曲線,所用電解質為O2飽和的KOH溶液,掃描速率為20mV/s,電勢掃描范圍為-0.6~0.2V (vs.Hg/HgO)。從圖4可以看出,兩種催化劑的峰電流密度和氧還原起始電勢都明顯高于同等條件下的碳粉,說明在堿性電解質中催化劑的氧還原催化活性強于碳粉。對比在N2和NH3氣氛中二次燒結La0.7Sr0.3MnO3前驅氧化物的循環伏安曲線,發現前者的氧還原起始電勢和峰電流密度都高于后者,說明前者的氧還原催化活性強于后者,同時也說明La0.7Sr0.3MnO3前驅體物相結構破壞會導致其催化活性位數目減少,最終造成氧還原催化活性降低。

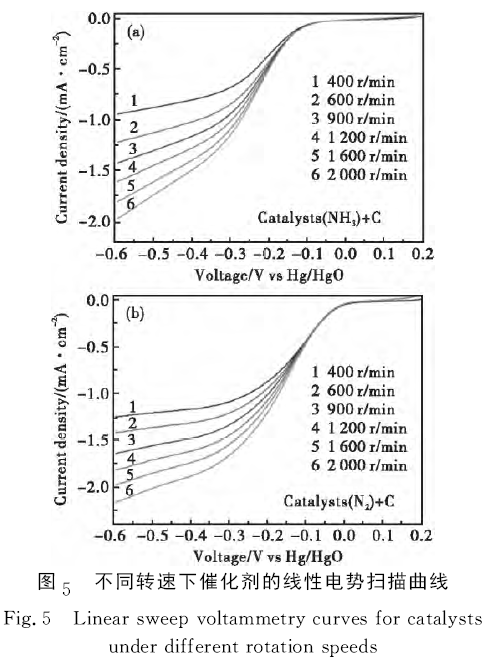
圖5為不同轉速下催化劑的線性電勢掃描曲線,所用電解質為O2飽和的KOH溶液,掃描速率為10mV/s,旋轉速度為400 r/min、600 r/min、900 r/min、1 200 r/min、1 600r/min和2 000r/min。由圖5可以看出,催化劑的極限電流隨轉速增加而增加。其中,NH3氣氛二次燒結催化劑樣品的極限電流密度由0.943mA·cm-2(400r/min)增至1.984mA·cm-2(2 000r/min),其氧還原起始電位為-0.062V (vs.Hg/HgO)。但N2氣氛二次燒結催化劑樣品的極限電流密度由1.247mA·cm-2(400r/min)提高到2.181mA·cm-2(2 000r/min),其起始電位為0.028V(vs.Hg/HgO)。N2氣氛二次燒結催化劑樣品的2個電化學參數均顯著高于NH3氣氛二次燒結催化劑樣品,說明前者的氧還原催化活性高于后者,但明顯低于La0.7Sr0.3MnO3前驅體[7],可能與二次燒結引起催化劑樣品顆粒晶體再生長有關。
3 結論
采用溶膠-凝膠法結合二次高溫燒結技術成功制備了錳系鈣鈦礦催化劑。XRD與EDS結果表明,N2氣氛二次燒結不改變La0.7Sr0.3MnO3前驅體物相,但NH3氣氛二次燒結會導致La0.7Sr0.3MnO3前驅體分解為(La0.7Sr0.3)2MnO4-δNδ(0≤δ<4)、MnO1-xNx和SrMnO2.694。電化學結果表明,N2氣氛二次燒結催化劑的氧還原催化活性高于NH3氣氛二次燒結催化劑,其氧還原起始電勢與極限電流分別為0.028V(vs.Hg/HgO)和2.181mA·cm-2(2 000r/min)。
參 考 文 獻
1 Yu Y,Li H,Wang H,et al.A review on performance degradationof proton exchange membrane fuel cells during startup and shutdownprocesses:Causes,consequences,and mitigation strategies[J].JPower Sources,2012,205:10.
2 Nrskov J K,Rossmeisl J,Logadottir A,et al.Origin of the overpotential for oxygen reduction at a fuel-cell cathode [J].J PhysChem B,2004,108(46):17886.
3 Fernandes A C,Paganin V A,Ticianelli E A.Degradation study ofPt-based alloy catalysts for the oxygen reduction reaction in protonexchange membrane fuel cells[J].J Electroanalytical Chem,2010,648(2):156.
4 Miyazaki K,Sugimura N,Matsuoka K,et al.Perovskite-type oxides La1-xSrxMnO3for cathode catalysts in direct ethylene glycol alkaline fuel cells[J].J Power Sources,2008,178(2):683.
5 Li D,Li S,Pan M.Study on the catalytic activity of perovskite-typeoxide La0.65Sr0.3MnO3for oxygen reduction[J].J Hubei University:Nat Sci Ed,2011,33(1):99(in Chinese).
李丹林,李賞,潘牧.鈣鈦礦型氧化物La0.65Sr0.3MnO3對氧還原的催化活性研究[J].湖北大學學報:自然科學版,2011,33(1):99.
6 Xu T,Wang G,Yu Y,et al.Study on the electrochemical and kinetic characteristics of La0.7Sr0.3MnO3[J].Chinese Battery Ind,2012,17(6):365(in Chinese).
徐甜,汪廣進,余意,等.La0.7Sr0.3MnO3電化學特征及氧還原動力學研究[J].電池工業,2012,17(6):365.
7 Wang G,Xu T,Wen S,et al.Structure-dependent electrocatalyticactivity of La1-xSrxMnO3for oxygen reduction reaction [J].SciChina Chem,2015,58(5):871.
8 Suntivich J,Gasteiger H A,Yabuuchi N,et al.Design principlesfor oxygen-reduction activity on perovskite oxide catalysts for fuelcells and metal-air batteries[J].Nat Chem,2011,3(7):546.
9 Wang Y,Cheng H P.Oxygen reduction activity on perovskite oxidesurfaces:A comparative first-principles study of LaMnO3,LaFeO3,and LaCrO3[J].J Phys Chem C,2013,117(5):2106.
10Tulloch J,Donne S W.Activity of perovskite La1-xSrxMnO3catalysts towards oxygen reduction in alkaline electrolytes[J].J PowerSources,2009,188(2):359.
11Xu T,Wang G,Liang C,et al.N-doped La2Zr2O7as an enhancedelectrocatalyst for oxygen reduction reaction[J].Electrochimica Acta,2014,143:83.
12 Atsumi T,Kamegashira N.Decomposition oxygen partial pressuresof Ln1-xSrxMnO3(Ln= La,Nd and Dy)[J].J Alloys Compd,1997,257(1):161.
13 Andrieux M,Picardf1 C.Nonstoichiometry and phase stability ofLa0.8Sr0.2MnO3±δat 1273K [J].J Mater Sci Lett,2000,19(8):695.